One of the most persistent mysteries in biology is why aging happens and how it unfolds differently across tissues and cells. After decades of studying aging through the lens of genetics, evolution, and molecular damage, we're proposing something fundamentally different: aging as a dissipative dynamical process, quantified through computational analysis of over 65 million cells.
Why Traditional Theories Fall Short
For over a century, scientists have grappled with explaining aging. August Weismann proposed in 1891 that aging was genetically programmed a sacrifice for the good of the species. Others suggested it was an evolutionary tradeoff, where genes beneficial in youth become harmful in old age. The free radical theory blamed accumulated molecular damage from oxidative stress. More recently, researchers have cataloged the "hallmarks of aging" genomic instability, telomere attrition, cellular senescence, and others.
Each theory captures part of the truth, but none provides a unifying framework. Why? Because they treat aging as a static endpoint rather than what it truly is: a dynamic process unfolding through time across multiple scales of biological organization.
Aging isn't merely the passage of time it reflects the evolving state of biological systems. The same chronological age can correspond to vastly different biological states depending on tissue type, cellular context, environmental exposures, and disease status. We need a framework that captures this complexity.
A New Framework: The Physics of Aging
What if we viewed biological organisms not just as collections of genes and proteins, but as dynamical systems complex networks of interacting components that evolve according to mathematical principles? This isn't metaphor; it's a rigorous mathematical framework borrowed from physics and applied to biology.
Using ergodic theory and the Hopf decomposition theorem, we can decompose any dynamical system into two fundamental components:
- Conservative Component: These are the recurrent, cyclic processes like circadian rhythms or cell cycle regulation that maintain homeostasis. In phase space (the space of all possible system states), conservative dynamics return to the same states over and over. They preserve structure, maintain order, resist entropy.
- Dissipative Component: These are the non-recurrent processes where the system drifts away from its initial configuration and never returns. Molecular damage accumulates, regulatory precision degrades, cellular organization becomes increasingly disordered. Entropy rises inexorably.
"In biological dynamical systems, what we understand as aging represents the predominance of dissipative forces which lead to deviation from recurrent states and increased entropy over time."
This is our central thesis: aging is fundamentally a dissipative process. Youth is characterized by strong conservative forces that maintain cellular order and functional integrity. Aging occurs when dissipative forces begin to dominate when repair mechanisms can no longer keep pace with damage, when regulatory networks lose precision, when the system drifts further from its optimal state.
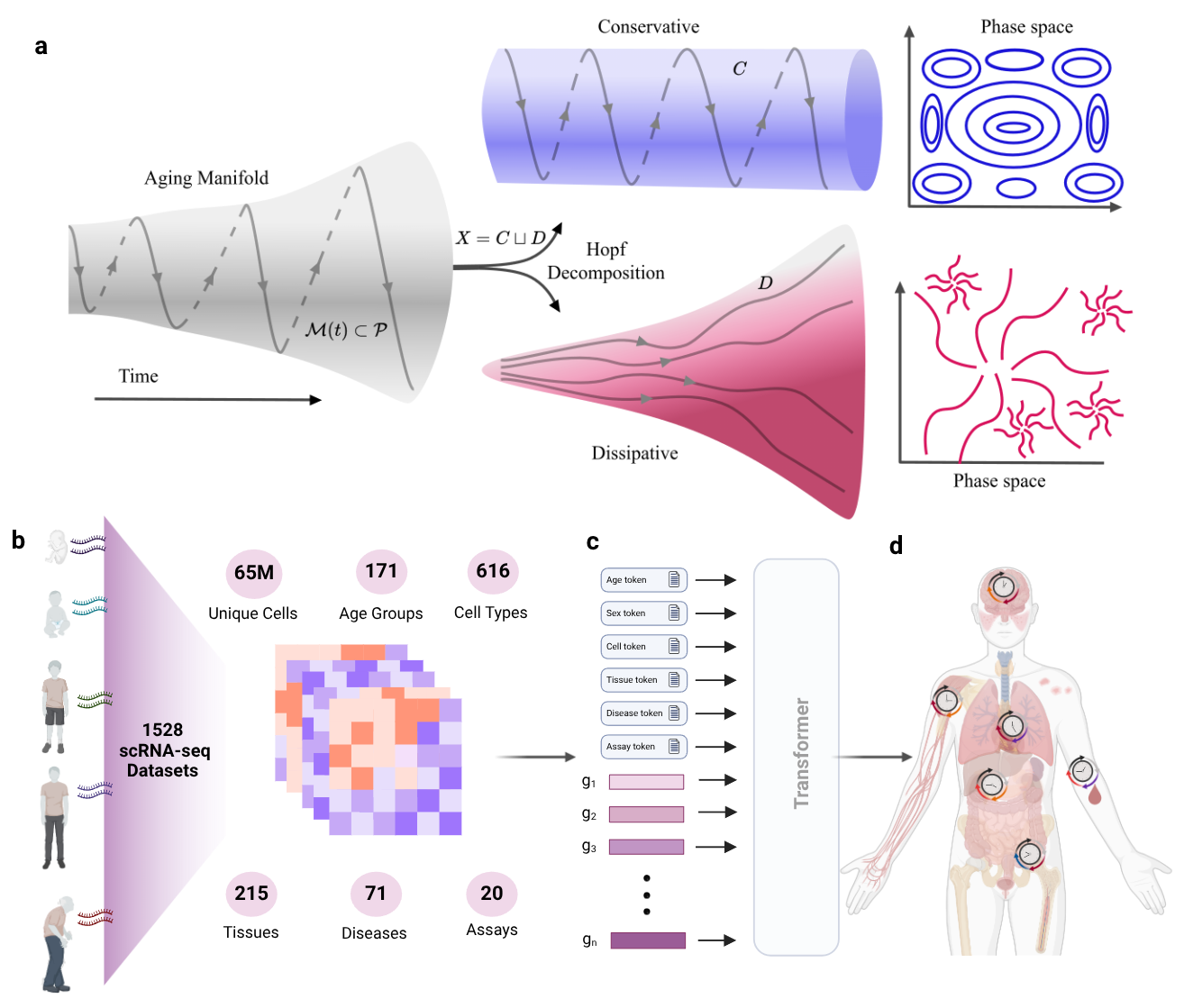
Figure 1: Overview of the theoretical and computational framework.
The Computational Revolution: 65 Million Cells Tell a Story
Theory means nothing without data. Traditional approaches to studying aging relied on candidate gene studies, model organisms, or small-scale transcriptomic analyses. We took a radically different approach: leverage the massive corpus of single-cell RNA sequencing data that has accumulated over the past decade.
Our dataset is staggering in scope:
- 65+ million individual cells analyzed
- 171 distinct age groups spanning from embryonic development to advanced old age
- 616 different cell types across 215 tissues
- 71 disease states enabling comparison of healthy versus pathological aging
This is a comprehensive dataset covering the full spectrum of human cellular diversity across the entire lifespan and multiple disease contexts. It's the kind of dataset that makes entirely new questions answerable.
Machine Learning Meets Dynamical Systems
Here's where it gets technically interesting. We can't directly observe the manifold of aging the high-dimensional space that captures all possible aging trajectories. It's too complex, too high-dimensional. But we can learn a representation of it.
We employed a transformer-based machine learning model the same architecture that powers large language models but trained on gene expression rather than text. The key insight: treat age as a token in the model's vocabulary. This allows the model to learn contextualized embeddings where genes, tissues, cell types, and ages are all represented in the same high-dimensional space.
These embeddings aren't just convenient data structures they're Lipschitz continuous representations of the underlying dynamical manifold. Small changes in biological state correspond to small changes in embedding space. This mathematical property means we can analyze aging dynamics by studying how embeddings drift and diverge over time.
Key Findings: The Cellular Aging Map
When we applied our model to construct the Cellular Aging Map (CAM), patterns emerged that fundamentally challenge how we think about chronological versus biological age.
1. Chronological Age Is Inadequate
Our model can predict molecular age from gene expression patterns. In many cases, predicted age closely matches chronological age suggesting that the aging process in those cells tracks linearly with time. But in many other cases, there are dramatic deviations.
Consider the respiratory airway: a Pearson correlation of 0.93 between predicted and chronological age. These cells age like clockwork, their molecular signatures evolving in lockstep with the calendar. This likely reflects the rapid turnover of airway cells they're constantly being replaced by stem cell populations, and the timescale of cellular turnover is so much faster than the timescale of dissipative aging.
Now contrast this with breast tissue: significant heterogeneity in age predictions, with some cells appearing molecularly much older or younger than their chronological age. This heterogeneity reflects the complex hormonal regulation, epigenetic remodeling, and differential exposures that modulate aging trajectories in these tissues.
The key insight: Age is contextual. Some cells are governed primarily by chronological time. Others are dominated by factors like inflammation, environmental exposures, hormonal cycling, or mechanical stress. Our model can distinguish between these modes.
2. Tissue-Specific and Cell-Type-Specific Aging
Not all tissues age equally. We quantified this by measuring the similarity between tissue/cell-type embeddings and the age token. High similarity means that tissue's molecular signature is strongly coupled to aging. Low similarity means resistance or delayed onset of molecular aging.
High-aging tissues: Yolk sac, breast, spleen, liver, bone marrow, prostate. These are high-turnover, metabolically active tissues in sync with systemic aging processes.
Low-aging tissues: Adrenal gland, eye, blood, esophagus. These show reduced susceptibility to molecular aging signals potentially reflecting protective mechanisms or unique aging trajectories.
At the cellular level, naive T cells and regulatory T cells showed high correlation with age exactly as we'd expect for cells generated by the thymus at rates that decline with age. They're molecular markers of immunological aging. In contrast, retinal rod cells and certain fibroblast populations showed low correlation context-driven cells influenced more by local microenvironment than systemic time.
3. Nonlinear Aging Dynamics
Perhaps most surprisingly, aging isn't linear. When we tracked the age gap (predicted minus chronological age) across the lifespan, we observed acceleration in early decades followed by deceleration after the fourth decade.
This nonlinearity has profound implications: different molecular processes dominate at different life stages. Early life is characterized by developmental programs and maturation. Middle age sees the emergence of cumulative damage and dysregulation. Late life involves complex compensatory mechanisms and system-wide reorganization. Any intervention targeting aging must account for this stage-specific heterogeneity.
The Molecular Choreography of Aging
Embeddings capture static snapshots and reveal temporal dynamics. By calculating cosine similarity between gene embeddings and age embeddings across different life stages, we can track how individual genes' roles evolve during aging.
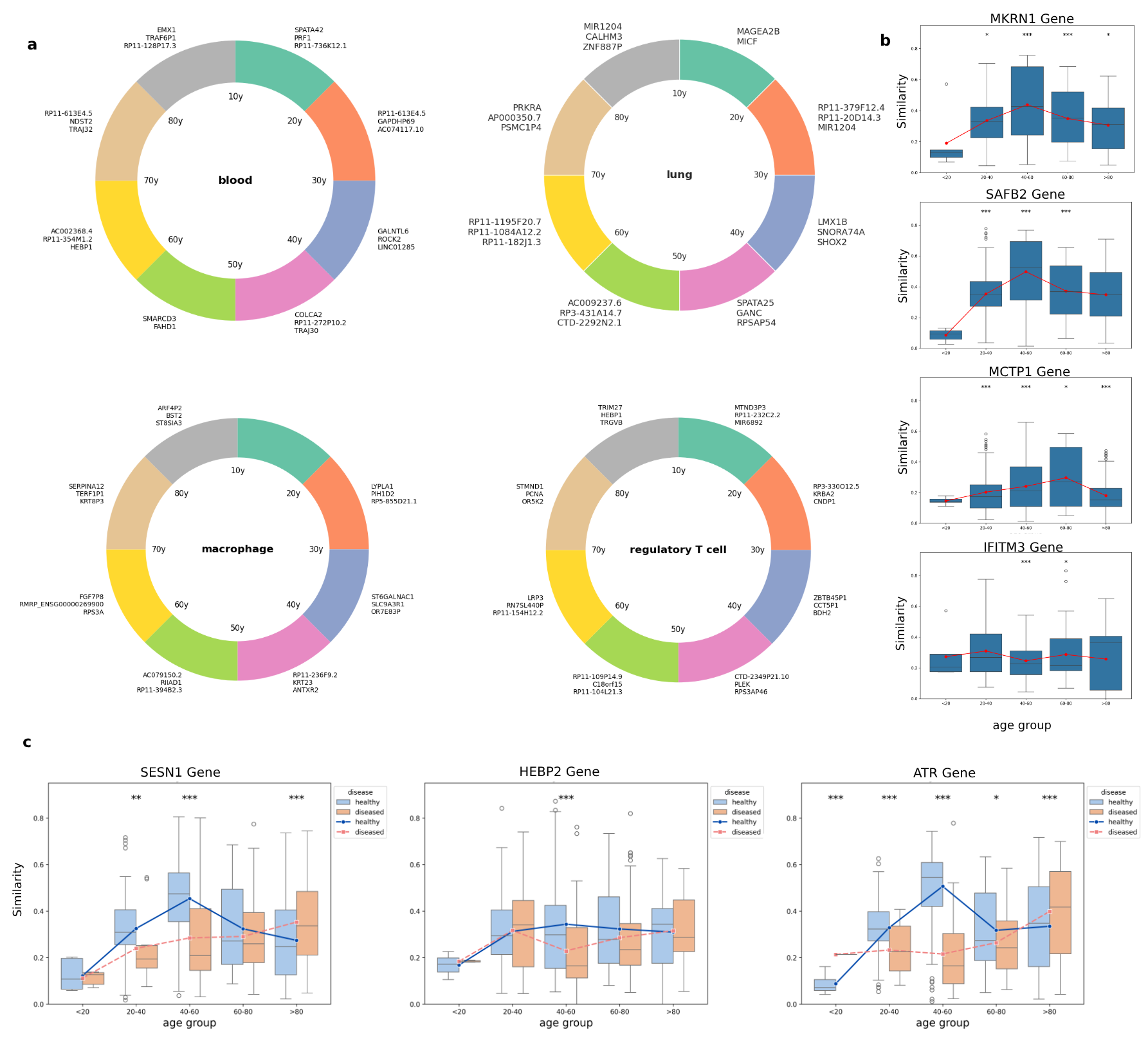
Figure 4: Gene embedding trajectories reveal stage-specific contributions to aging. (a) Different genes dominate at different life stages in blood, lung, macrophages, and regulatory T cells. (b) Some genes like IFITM3 show stable similarity across lifespan, while others exhibit peaked or transient interactions. Disease alters these trajectories genes like ATR show diminished association during adulthood in disease but overcompensation in old age, suggesting impaired DNA repair followed by maladaptive response.
Stage-Specific Gene Contributions
In blood tissue, genes like SPATA42 and PRF1 have their highest impact during adolescence. But between ages 70-80, completely different genes TRAJ32 and NDST2 dominate the aging signature. This isn't random noise; it reflects the biological reality that aging involves sequential waves of molecular changes.
Some genes, like IFITM3 (involved in antiviral defense), maintain relatively stable similarity across the lifespan constitutive housekeeping functions that don't drift much with age. Others show sharply peaked interactions at specific time points. MKRN1 and SAFB2 spike during early to mid-life, suggesting regulatory roles during developmental or reproductive maturation. MCTP1 shows increasing similarity with later age embeddings, potentially reflecting involvement in age-associated stress responses.
Disease Rewrites the Aging Program
When we compared gene trajectories in healthy versus diseased aging, the patterns were striking. Genes like SESN1, HEBP2, and ATR showed complex temporal profiles in healthy samples but fundamentally altered trajectories in disease.
ATR a critical DNA damage response gene showed diminished association during adulthood in diseased samples, suggesting early impairment of DNA repair. But in old age, diseased samples showed higher ATR similarity than healthy ones. This is consistent with a two-phase response: initial failure of protective mechanisms, followed by compensatory overexpression that may itself be maladaptive.
These shifts demonstrate that age-related diseases don't just accelerate normal aging they fundamentally disrupt the coordinated temporal regulation of protective gene programs.
Dissipative and Conservative Genes: The Heart of the Theory
Now we arrive at the core empirical test of our theory. If aging is indeed a dissipative process, we should be able to identify genes that behave conservatively (maintaining stable embeddings over time) versus dissipatively (showing significant temporal drift).
We quantified this by measuring maximum embedding drift for each gene how far its embedding can move from baseline across the entire age spectrum. Then we validated statistical significance using permutation tests, shuffling age labels 1,000 times to build null distributions.
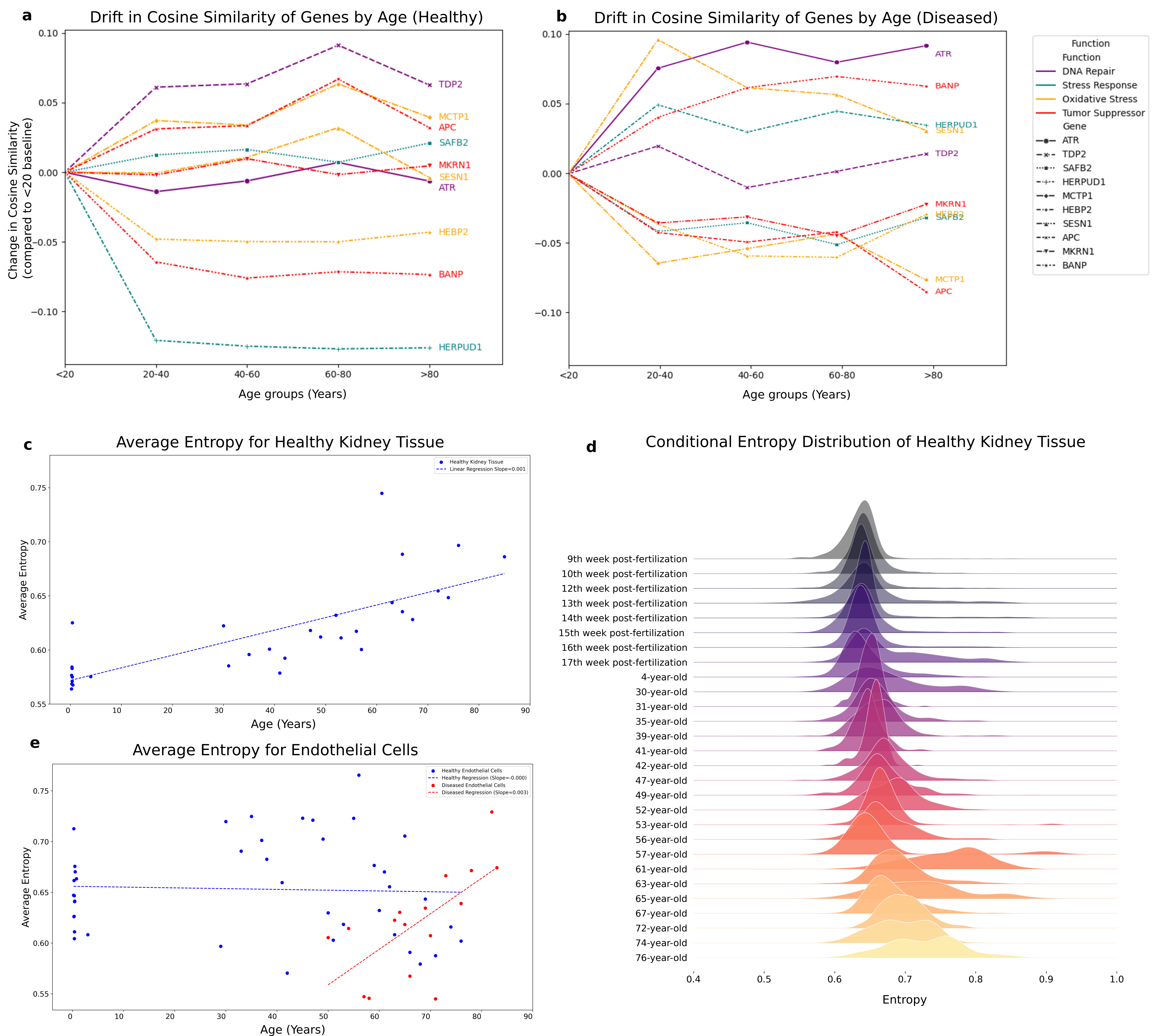
Figure 5: Dissipative versus conservative genes. (a) In healthy tissue, genes like MKRN1, SESN1, and ATR show minimal drift (conservative), while ALDH3B1, NR2C2, and HERPUD1 exhibit high drift (dissipative). (b) Disease transforms gene behavior ATR becomes dissipative while TDP2 becomes conservative. (c,d) Permutation testing validates dissipative genes with mean Z-score = 5.37 and adjusted p-value = 0.005. (e) Phase portrait showing dissipative genes cluster by divergence and recurrence metrics.
Conservative Genes: The Guardians of Homeostasis
Genes like MKRN1, SESN1, and ATR were classified as conservative their embeddings remained stationary across all life stages. These genes maintain stable dynamics throughout aging, suggesting fundamental roles in cellular homeostasis that must be preserved for survival.
ATR is particularly interesting: it's a master regulator of DNA damage response and cell cycle checkpoints. Its conserved behavior makes sense you can't afford to have DNA repair mechanisms drifting all over the place as you age. They must remain stable and functional, or else genomic instability spirals out of control.
Dissipative Genes: The Harbingers of Decline
In contrast, genes like ALDH3B1 (aldehyde dehydrogenase), NR2C2 (nuclear receptor), and HERPUD1 (ER stress response) showed high temporal drift. These dissipative genes capture processes that are dynamically reshaped during aging metabolic rewiring, stress responses, regulatory network reorganization.
Crucially, the distinction between conservative and dissipative appears independent of biological function. Both categories include genes involved in DNA repair, stress response, metabolism, and regulation. This universality suggests that conservation and dissipation are intrinsic characteristics of the aging process itself, not tied to specific pathways.
Disease Transforms Gene Classification
Here's where it gets really interesting: disease can flip genes between categories.
ATR, conservative in healthy cells, became markedly dissipative in diseased samples. This suggests that disease destabilizes even the most fundamental maintenance systems. When DNA repair mechanisms start to drift, genomic instability accelerates, mutations accumulate, and cellular function deteriorates.
Conversely, TDP2 a dissipative gene in health became conservative in disease. This counterintuitive finding might reflect a disease-specific constraint that locks certain genes into particular expression states, eliminating the normal variability they exhibit in healthy aging.
Statistical Validation
Our permutation tests confirmed the robustness of this classification. Dissipative genes showed drift scores in the extreme positive tail of their null distributions (mean Z-score = 5.37, with 65% showing Z > 3). After Benjamini-Hochberg correction for multiple testing, the mean adjusted p-value was 0.005 highly significant.
Conservative genes, as expected, showed minimal statistically significant temporal drift. The phase portrait analysis further demonstrated that dissipative genes cluster distinctly in divergence-recurrence space, validating our dynamical systems classification.
Entropy: Quantifying the Loss of Order
Entropy has long been proposed as a conceptual framework for understanding aging the progressive loss of molecular order and information. But how do you actually measure it? Previous work lacked concrete metrics to quantify information loss systematically.
We used Shannon entropy calculated from our model's predictions. When the model masks a token (say, a specific tissue type), it outputs a probability distribution over all possible tissue types. High entropy in this distribution means high uncertainty the model can't confidently predict what's been masked. Low entropy means confident, specific predictions.
By conditioning these predictions on age, we can track how molecular disorder evolves across the lifespan. Higher entropy indicates greater uncertainty and loss of molecular specificity the hallmark of dissipative aging.
Two Patterns of Entropic Aging
When we analyzed entropy dynamics across tissues and cell types, two distinct patterns emerged:
Pattern 1: Progressive Entropy Increase (Kidney Cells)
Healthy kidney cells showed exactly what you'd expect from classical aging theory: entropy increases progressively with age, with the rate of increase accelerating in later life. The distribution of entropy across age groups showed marked increases in variation heightened heterogeneity potentially signaling either greater molecular uncertainty or increased cellular plasticity.
This is dissipation in its purest form: continuous, irreversible loss of molecular organization. No recovery, no compensation just relentless drift toward disorder.
Pattern 2: Episodic Entropy Spikes (Endothelial Cells)
Endothelial cells told a completely different story. No consistent trend of entropy increase across aging. Instead, sharp but transient entropy spikes at specific life stages mid-thirties, early fifties, early sixties followed by subsequent reductions.
These "entropy spikes" suggest episodic disruptions in molecular organization that are later buffered, stabilized, or even repaired. This points to a more nuanced view: aging as a dynamic balance between perturbation and recovery, decline and compensation. Endothelial cells retain adaptive capacity to respond to local instabilities.
This makes biological sense. Endothelial cells line blood vessels they're constantly exposed to mechanical forces, inflammatory signals, and metabolic stress. They need resilience. Their native state is uncommitted and elastic, capable of profound adaptation. This elasticity only erodes when disease dominates.
Disease Disrupts Entropic Compensation
Disease-transformed endothelial cells consistently exhibited higher entropy than healthy counterparts demonstrating sustained loss of molecular specificity and increased structural disorder. The adaptive recovery mechanisms present in healthy aging appear overwhelmed or disabled in disease states.
This suggests that disease and aging interact synergistically. Disease doesn't just accelerate normal aging it compounds molecular disorganization and eliminates compensatory mechanisms. The combination of disease-related stress and age-associated deterioration drives continuous entropy elevation, reducing resilience and functional capacity.
Implications: From Theory to Therapy
What does all this mean for how we understand and potentially intervene in aging?
1. Age Is Contextual, Not Universal
Chronological age is a crude proxy for biological state. Some cells and tissues track chronological time faithfully (respiratory airway, naive T cells), while others are dominated by inflammation, mechanical stress, hormonal regulation, or environmental exposures (breast tissue, certain fibroblasts).
This has profound implications for "anti-aging" interventions. Therapies must be tissue-specific and context-dependent. A one-size-fits-all approach will fail because different tissues age through different mechanisms at different rates.
2. Identify and Target Dissipative Genes
Dissipative genes represent vulnerabilities points where the system is losing stability and drifting toward disorder. But they're also potential intervention targets. If we can understand what drives their temporal drift epigenetic changes, loss of regulatory precision, accumulated damage we might be able to slow or reverse the dissipative process.
Conservative genes, meanwhile, represent resilience core functions that must be preserved for survival. Protecting these genes from disease-induced destabilization could prevent the catastrophic loss of homeostatic capacity that characterizes pathological aging.
3. Harness Endogenous Recovery Mechanisms
The episodic entropy dynamics in endothelial cells reveal something crucial: some tissues possess endogenous mechanisms to recover from perturbations. These aren't passive victims of aging they're active systems capable of responding, adapting, and compensating.
Understanding these recovery mechanisms could enable therapies that enhance resilience rather than just slowing decline. Instead of fighting aging head-on, we might be able to amplify the body's own capacity for molecular reorganization and repair.
4. Stage-Specific Interventions
Aging is nonlinear, with different molecular processes dominating at different life stages. Early life involves developmental programs and tissue maturation. Middle age sees the emergence of cumulative damage and regulatory drift. Late life is characterized by system-wide reorganization and compensatory changes.
This means interventions must be stage-specific. What works in middle age (preventing initial damage) may be ineffective or even harmful in old age (where the priority is maintaining compensatory mechanisms). Precision medicine for aging must account for where someone is on their aging trajectory.
5. Disease Disrupts Aging But How?
Disease doesn't just accelerate aging it fundamentally alters aging trajectories. Conservative genes become dissipative. Dissipative genes become locked into pathological states. Compensatory mechanisms fail. Entropy spirals upward without recovery.
This suggests that preventing age-related diseases isn't just about avoiding specific pathologies it's about preserving the fundamental dynamical properties that enable healthy aging. When disease disrupts the balance between conservative and dissipative forces, aging accelerates catastrophically.
Limitations and Future Directions
No framework is perfect, and ours has limitations we must acknowledge.
Interpretability: Embeddings are powerful but abstract. While we can track drift and measure entropy, direct mechanistic interpretation remains challenging. Complementary experimental approaches will be needed to validate causal mechanisms.
Dataset biases: Despite our comprehensive data, certain tissues, cell types, and age ranges remain underrepresented. Generalizability requires continued expansion of single-cell atlases across diverse populations and conditions.
Transcriptional focus: We've analyzed gene expression, but aging involves epigenetic, proteomic, metabolomic, and structural changes. Multi-omics integration will provide a more complete picture.
Model vs. data uncertainty: High entropy could reflect true biological variability or model uncertainty. Distinguishing these requires careful experimental design and validation.
Temporal resolution: Our analysis captures gradual changes across age bins, but some aging processes may involve rapid transitions or transient states we're missing. Higher temporal resolution studies particularly longitudinal tracking of individuals will be crucial.
A New Paradigm
For decades, aging research has been fragmented geneticists studying longevity genes, cell biologists examining senescence, epidemiologists tracking age-related diseases. Each discipline generated insights, but lacked a unifying framework.
By framing aging as a dissipative dynamical process and providing computational tools to quantify it, we offer a path toward integration. Aging isn't random decay, genetic programming, evolutionary tradeoff, or damage accumulation in isolation. It's the progressive dominance of dissipative over conservative forces quantifiable through temporal drift, entropy production, and phase space dynamics.
This framework doesn't replace existing theories; it subsumes them. Damage accumulation drives dissipation. Genetic programs establish initial conservative structures. Evolution shapes the balance between maintenance and reproduction. But all of these operate within a dynamical system that can be rigorously analyzed and potentially modulated.
Conclusion: Toward Rational Interventions
The ultimate test of any biological theory is whether it enables intervention. Can we use this framework to actually slow aging or ameliorate age-related decline?
Our cellular aging map provides unprecedented resolution to identify:
- Which tissues are most susceptible to dissipative aging
- Which genes transition from conservative to dissipative behavior
- When during the lifespan these transitions occur
- How disease disrupts normal aging trajectories
- Which systems retain capacity for recovery and compensation
Armed with this knowledge, we can begin designing rational interventions: tissue-specific therapies, stage-specific treatments, strategies to preserve conservative genes, approaches to slow dissipative drift, methods to enhance endogenous recovery mechanisms.
The future of aging research lies in viewing it as a dynamic process we can measure, understand, and potentially modulate. By understanding the forces that drive systems toward disorder, we create the possibility of opposing them.
Read the full paper: https://doi.org/10.1038/s41514-025-00277-2