How do thousands of genes working together give rise to the remarkable diversity of cells, tissues, and organisms we observe in nature? This question the genotype-phenotype relationship has captivated biologists since Wilhelm Johannsen first coined these terms in 1909. A century later, we're still grappling with how to precisely map this connection. But now, we have new tools.
The Genotype-Phenotype Problem
Understanding how genetic information (genotype) translates into observable characteristics (phenotype) remains one of biology's grand challenges. Single-cell RNA sequencing (scRNA-seq) has revolutionized our ability to measure gene expression at cellular resolution, revealing the complex dynamics of thousands of genes simultaneously. Yet this technological leap has created its own challenge: how do we make sense of these massive, high-dimensional datasets?
Traditional approaches forward genetics (gene to phenotype) and reverse genetics (phenotype to gene) fall short when confronted with the scale and complexity of modern genomic data. In human cells, thousands of genes interact to produce an incredibly diverse phenome landscape. The sheer combinatorial space is overwhelming. Moreover, gene expression isn't just determined by cellular identity it's highly responsive to age, sex, tissue context, developmental stage, and disease state.
Current machine learning models, while powerful, often lack interpretability. They focus on resolving cellular heterogeneity but miss the multifaceted nature of gene expression patterns. They treat genes in isolation from their biological context, failing to capture how the same gene can play different roles in different tissues, cell types, or physiological conditions.
Integrated Genetics: A New Paradigm
We propose a fundamentally different approach: integrated genetics. Instead of studying genotype and phenotype separately, we learn them simultaneously using self-supervised language models. Our method, embodied in a foundation model we call PolyGene, integrates gene expression data with rich phenotypic metadata tissue type, cell type, age, sex, disease state to create contextualized representations of the genotype-phenotype relationship.
The key insight: treat biological data like natural language. Each gene is like a word, each cell state like a sentence. But unlike previous models that simply rank genes by expression level, we embed genes with their biological context. This allows the model to learn not just what genes are expressed, but where, when, and under what conditions they're expressed.
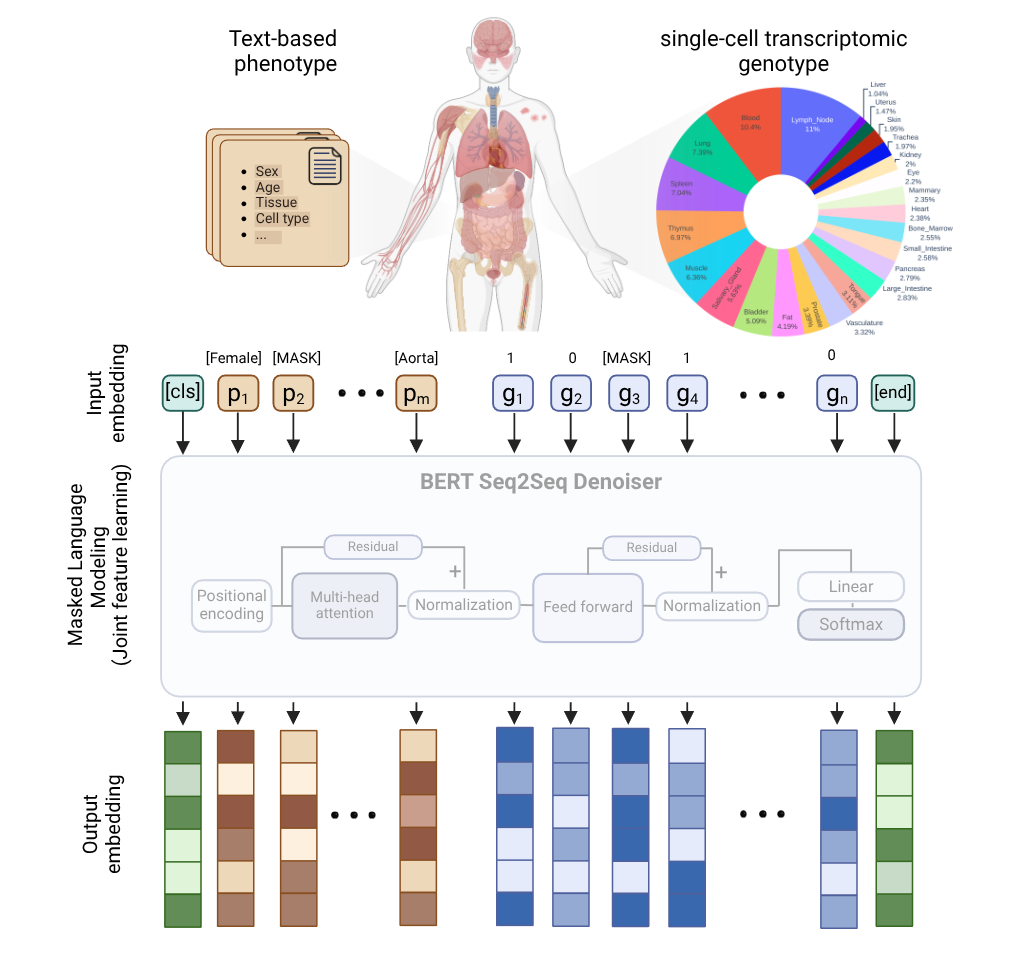
Figure 1: Overview of the PolyGene model. Gene expression data from single-cell RNA-seq and associated phenotypes (tissue, cell type, age, sex) are embedded as input to a transformer-based language model. The model learns the genotype-phenotype relationship in an integrated manner through masked language modeling randomly masking both genes and phenotypes during training. The outputs are high-dimensional embeddings that capture rich contextual relationships between genes and their biological contexts.
The PolyGene Foundation Model
We trained PolyGene on the Tabula Sapiens cell atlas nearly 500,000 human cells across 24 organs from 14 donors spanning various age ranges. This comprehensive dataset provides unprecedented coverage of human cellular diversity under healthy conditions.
Architecture and Innovation
PolyGene is built on a transformer architecture similar to BERT, but with crucial innovations for biological data:
- Fixed positional encoding: Unlike previous models that order genes by expression level (which is sensitive to noise), we use consistent positional embeddings for each gene regardless of expression, with padding for unexpressed genes.
- Multimodal integration: Phenotypic metadata (tissue, cell type, age, sex) are embedded as learnable tokens alongside gene expression values.
- Differential masking: We mask phenotype tokens at 50% probability versus 15% for gene tokens, forcing the model to learn robust phenotype-genotype associations even with incomplete information.
- Soft labeling: Gene expression is binned, and we use soft labels that spread probability across adjacent bins, improving convergence and capturing the ordinal nature of expression levels.
The model learns through masked language modeling: randomly mask portions of the input (both genes and phenotypes), then train the model to reconstruct what's been masked based on context. This self-supervised approach requires no labeled data beyond the metadata that already exists in single-cell datasets.
Resolving Cellular Heterogeneity with Context
One of the primary challenges in single-cell analysis is cellular heterogeneity the fact that even "identical" cell types can show substantial variation. Where does this variation come from? Technical noise? Biological diversity? Developmental stage? Disease state?
By integrating phenotypic metadata directly into the learning process, PolyGene produces embeddings that naturally account for sources of variation. These contextualized embeddings dramatically improve our ability to distinguish cell types, tissue origins, donor age, and sex.
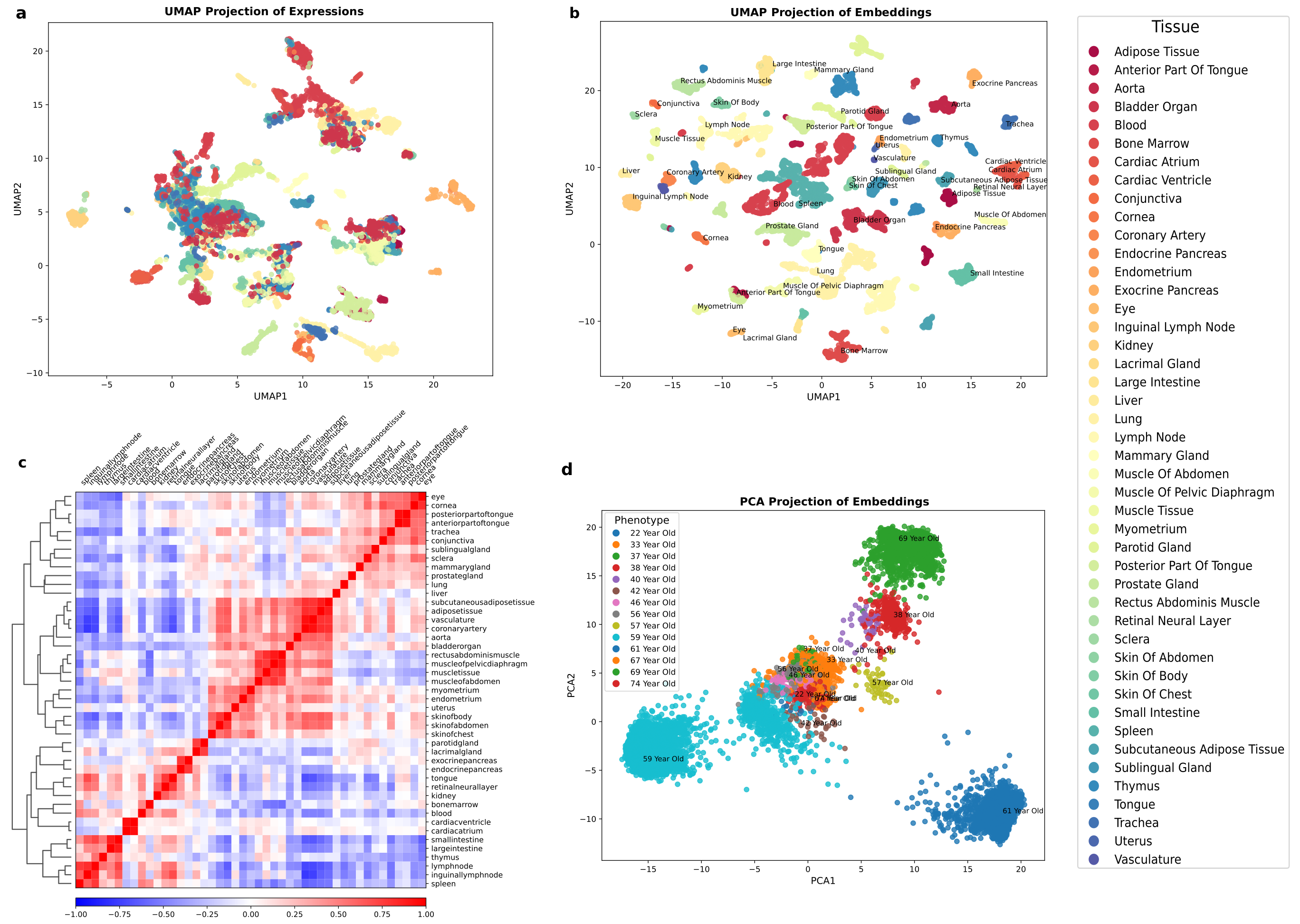
Figure 2: Resolving heterogeneity in phenotypes using contextualized embeddings. (a) UMAP visualization of the human cell atlas colored by tissue type using standard unsupervised clustering on gene expression shows overlapping tissue groups. (b) The same cells visualized using information-rich embeddings from PolyGene's output reveal clearer tissue separation and identification of subclusters within several tissues including thymus, trachea, subcutaneous adipose, and parotid gland. (c) Correlation analysis of tissue embeddings demonstrates hierarchical organization aligned with biological relationships cardiac ventricle and atrium show high similarity, while unexpected connections like tongue-kidney similarity align with known cross-tissue biomarkers for chronic kidney disease. (d) Linear dimension reduction on age embeddings cleanly separates different age groups.
Cross-Tissue Biomarkers and Unexpected Connections
When we analyzed the similarity of tissue embeddings learned by PolyGene, fascinating patterns emerged. As expected, functionally related tissues like the cardiac ventricle and atrium showed high similarity. But we also discovered unexpected connections.
The tongue, retinal neural layer, and kidney showed surprisingly high similarity in their molecular signatures. Initially counterintuitive, this finding actually aligns with clinical observations: tongue diagnostics have proven relevant for detecting chronic kidney disease. This suggests PolyGene is capturing cross-tissue biomarkers molecular signatures that reflect systemic processes affecting multiple organs simultaneously.
We validated PolyGene's performance on independent datasets not seen during training over 75,000 cells from lung, liver, and blood studies. PolyGene consistently outperformed state-of-the-art methods like scGPT in precision, recall, and F1 scores for both tissue and cell-type classification, particularly excelling at distinguishing closely related cell types and states.
Context-Dependent Gene Functions
Here's where integrated genetics reveals something profound: genes don't have fixed meanings. The same gene can play entirely different roles depending on biological context tissue type, cell state, developmental stage, disease condition.
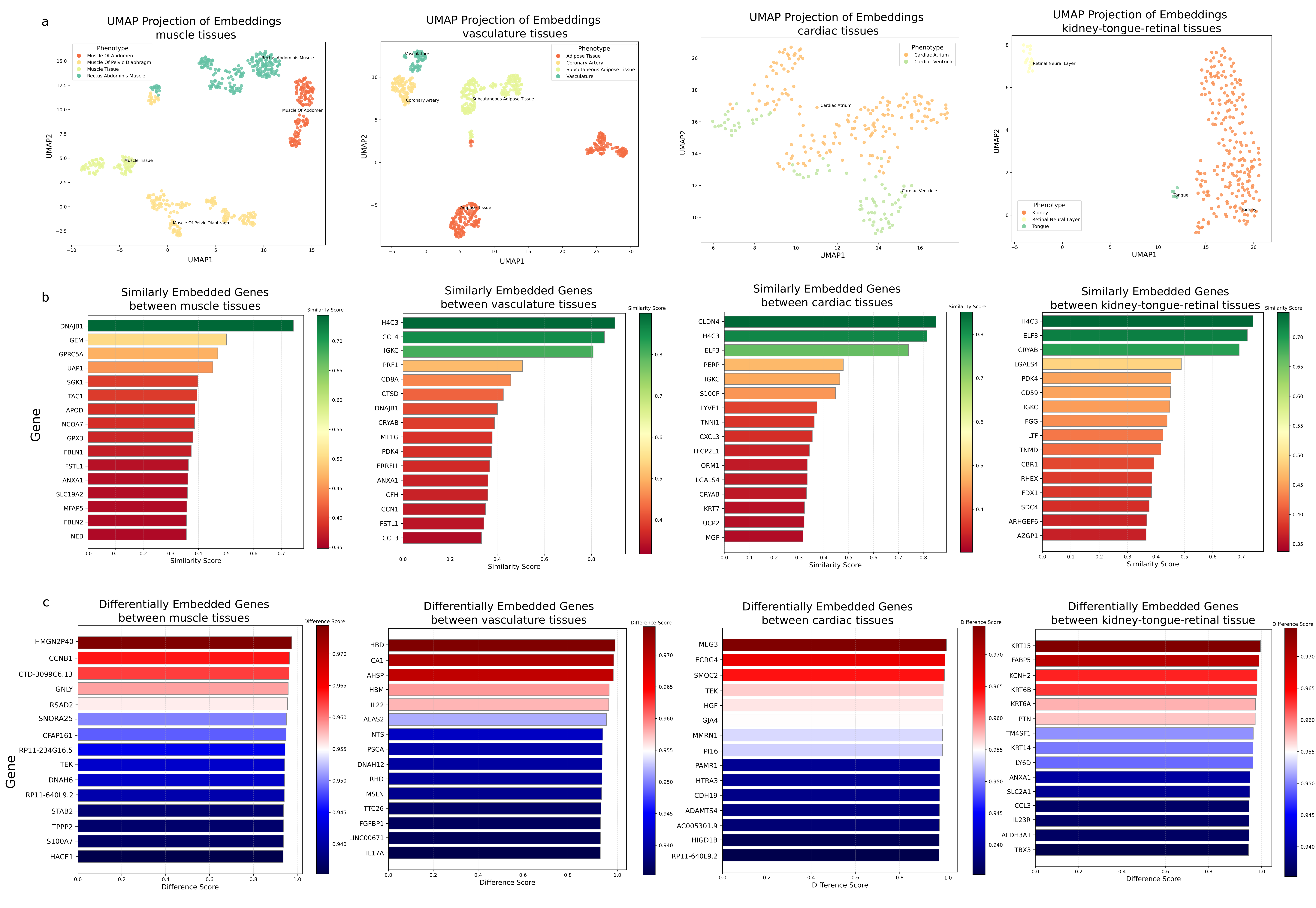
Figure 3: Obtaining contextualized genotype-phenotype relationships from embeddings. (a) UMAP visualization of tissue phenotype embeddings shows how tissues cluster by similarity muscle, vasculature, and cardiac tissues group together while kidney, tongue, and retinal tissue form distinct clusters. (b) Cosine similarity analysis between tissue embeddings and gene embeddings identifies genes with similar roles across these tissues. (c) Differentially embedded genes (DEGs) analysis reveals genes that show distinct embedding patterns across contexts such genes may have tissue-specific or context-dependent functions.
The IL-2 Example: Context Reshapes Function
Consider IL2 (Interleukin-2), a crucial immune signaling molecule. When we clustered tissues based on their IL2 gene embeddings, we found an unexpected pattern: inguinal lymph nodes, bone marrow, and skin of the chest/abdomen showed high similarity but were distinct from other lymph nodes and skin regions across the body.
This suggests IL2 plays multifaceted roles across tissues, potentially inducing similar immune responses in T and B lymphocytes at specific anatomical locations. The IL2 embeddings also clearly separated large and small intestines consistent with mouse studies showing IL2 is necessary for immunologic homeostasis in the large intestine but not the small intestine, indicating tissue-specific regulation.
KRT8: A Contrasting Pattern
In contrast, KRT8 (Keratin 8) embeddings showed small and large intestines clustering together a more uniform pattern aligning with traditional tissue similarity. This comparison illustrates that gene function heterogeneity isn't universal; some genes maintain consistent roles across related tissues while others adapt to local contexts.
Quantifying Context-Dependence
We systematically examined how gene embeddings vary across tissues and cell types. For genes like NICOA7, XBP1, BNIP3L, and CTSD, the embeddings showed closer proximity between aorta and subcutaneous adipose than between aorta and spleen despite all three tissues expressing these genes. This indicates context-dependent functional roles.
Conversely, genes like TIMP1, CD55, and HSPA5 showed minimal embedding variation across tissues, suggesting more uniform functions. At the cellular level, FBXO7 showed closer embeddings between memory B cells and naive B cells than between memory B cells and kidney epithelial cells exactly what we'd expect given functional similarity.
Aging Rewrites the Gene Network
One particularly striking application of PolyGene is analyzing how aging alters gene network structure. We focused on endothelial cells the cells lining blood vessels that are critical for vascular health and disease.
Gene interaction networks are known to exhibit scale-free architecture, where most genes have few connections but a small number of "hub" genes have many connections. This structure makes networks robust yet efficient. But how does aging affect this architecture?
Our analysis revealed that aging fundamentally restructures the gene network in endothelial cells. The power-law distribution of node connectivity changes, particularly affecting low-degree nodes specialized genes with fewer connections. These specialized nodes begin forming more connections during aging, diluting their specialized roles and potentially contributing to loss of cellular function.
Specific genes like KCNH8 (a potassium channel), DNAJA4 (a heat shock protein), and EGLN3 (a hypoxia-inducible factor) showed pronounced changes in their network embeddings with age, making them candidates for further investigation into vascular aging mechanisms.
Polyfunctional Genes: One Gene, Many Roles
Perhaps the most conceptually important discovery enabled by PolyGene is what we call gene polyfunctionality the ability of a single gene to play multiple, functionally distinct roles even within the same cell type.
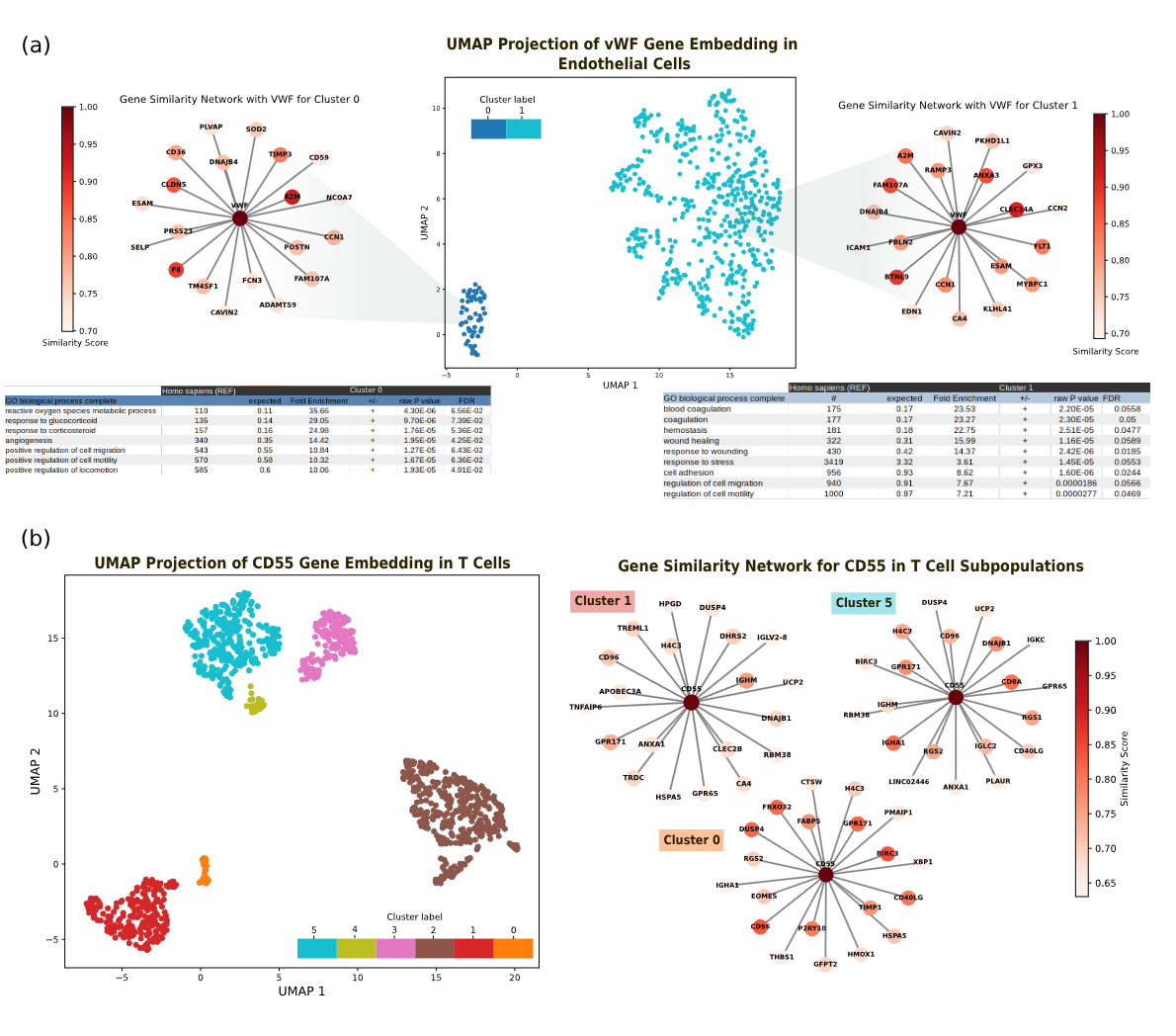
Figure 6: Polyfunctional characteristics revealed through embedding analysis. (a) Clustering of VWF (von Willebrand Factor) gene embeddings in endothelial cells reveals two distinct functional clusters. Cluster 0 shows VWF associated with reactive oxygen species metabolic processes and cellular stress responses a lesser-known function. Cluster 1 captures VWF's canonical role in blood coagulation and hemostasis. Both clusters exist within the same cell type, indicating context-dependent functional states. (b) CD55 embeddings in T cells reveal six distinct clusters, each potentially representing different functional contexts or regulatory states. Gene ontology enrichment for each cluster confirms distinct biological processes, demonstrating how embeddings can decode multifunctional gene roles.
The VWF Story: Two Functions, One Gene
We investigated VWF (von Willebrand Factor) in endothelial cells. VWF is famously known for its role in blood coagulation it's critical for hemostasis, helping platelets stick to damaged blood vessel walls.
But when we clustered VWF embeddings from different cellular contexts, two functionally distinct clusters emerged not separated by tissue, age, or any obvious phenotypic variable, but by molecular context:
- Cluster 1: The canonical blood coagulation function. Gene ontology enrichment confirmed this cluster involves hemostasis, platelet activation, and wound healing pathways.
- Cluster 0: A lesser-known function involving reactive oxygen species management and cellular response to glucocorticoid stress. This cluster suggests VWF plays a role in oxidative stress responses beyond its coagulation function.
This is polyfunctionality: the same gene, in the same cell type, adopting different functional roles depending on cellular state, signaling environment, or regulatory context.
CD55: A Six-Cluster Story
CD55, known for regulating the complement system in immune cells, showed even more dramatic functional diversity. In T cells, we identified six distinct embedding clusters, each associated with different biological processes and molecular pathways. This suggests CD55 operates in at least six functionally distinct contexts within T cells.
Not All Genes Are Polyfunctional
Importantly, not every gene exhibits polyfunctionality. When we analyzed KLF5 in endothelial cells, no distinct functional clusters emerged its embeddings formed a relatively homogeneous distribution, suggesting a more uniform role across contexts.
Implications for Drug Discovery
Polyfunctionality has profound implications for therapeutic development. If a gene plays multiple distinct roles within the same tissue or cell type, drugs targeting that gene might have context-dependent effects. By understanding which functional cluster is relevant to a disease state, we could design interventions that selectively modulate one function while leaving others intact.
This precision targeting specific functional states of genes rather than genes themselves could reduce side effects and improve therapeutic specificity.
Universal Phenotype Associations
Beyond tissue and cell-type specificity, we analyzed which genes most strongly associate with each phenotype across all contexts.
Universal genes: H4C3 (H4 clustered histone 3) showed the highest similarity to all phenotypes. This histone gene regulates chromatin structure and is involved in DNA repair, replication, and cell cycle progression fundamental processes present in all cells. Its universal prominence makes biological sense.
Age-associated genes: FST (Follistatin) emerged as highly similar to age embeddings. Follistatin regulates muscle growth, wound healing, and reproduction processes intimately connected to aging. Recent studies have shown follistatin increases muscle size and improves lifespan in mice, validating its relevance to aging biology.
Differentially embedded genes: We identified genes showing the most variation across phenotypes:
- Tissue: IGHV3-7, CCL17, IGKV6D-21 (immunoglobulin and chemokine genes)
- Cell type: CCL3, CA1, CCL4L2 (chemokines and carbonic anhydrase)
- Age: RGS1, CA1, CCL3 (G-protein signaling and immune genes)
- Sex: CA1, CCL17, EREG (metabolic and growth factor genes)
The prominence of immune-related genes across all phenotypic categories highlights the central role of immune system regulation in biological diversity and aging.
Implications and Future Directions
Integrated genetics through PolyGene offers several transformative capabilities:
1. Contextualized Gene Function
By embedding genes with their biological context, we move beyond static annotations ("Gene X does Y") to dynamic, context-dependent understanding ("Gene X does Y in tissue A under condition B, but does Z in tissue C under condition D"). This matches biological reality far better than traditional functional annotation.
2. Cross-Tissue Biomarkers
The unexpected tissue similarities revealed by PolyGene (tongue-kidney, for example) point toward systemic biomarkers molecular signatures that could enable non-invasive diagnosis of organ-specific diseases through accessible tissues like blood or saliva.
3. Polyfunctionality in Drug Discovery
Understanding that genes have multiple functional states within the same cell type enables unprecedented therapeutic precision. We can potentially target specific functional states relevant to disease while sparing others, reducing side effects and improving efficacy.
4. Network Medicine and Aging
The ability to track how gene networks restructure during aging or disease progression provides targets for intervention. Genes whose network positions change dramatically may represent vulnerabilities or opportunities for therapeutic manipulation.
5. Foundation for Personalized Medicine
PolyGene can be fine-tuned for specific downstream applications disease diagnosis, drug response prediction, cell type annotation, trajectory inference. Its contextualized representations provide a rich starting point for transfer learning to specialized tasks.
Limitations and Challenges
No approach is without limitations:
Computational demands: Training large foundation models requires substantial computational resources, potentially limiting accessibility for smaller research groups.
Data completeness: While Tabula Sapiens is comprehensive, it focuses on healthy tissues from limited donors. Expanding to diseased states, developmental stages, and diverse populations will improve generalizability.
Interpretability: While embeddings are powerful, translating high-dimensional representations back to mechanistic biological insights remains challenging. Complementary experimental validation is essential.
Phenotype coverage: We integrate tissue, cell type, age, and sex, but many other relevant phenotypes exist disease state, drug exposure, circadian time, nutritional status. Future iterations should expand phenotypic coverage.
A New Era for Genetics
Wilhelm Johannsen sought an "exact" description of the genotype-phenotype relationship in 1909. His vision was ahead of its time the tools didn't exist. Now, with single-cell genomics and modern machine learning, we're finally developing the computational frameworks to realize that vision.
Integrated genetics represents a paradigm shift: instead of studying genes and phenotypes separately, we learn them jointly, capturing their mutual dependence and contextual variability. PolyGene demonstrates this approach can resolve cellular heterogeneity, discover cross-tissue biomarkers, reveal context-dependent gene functions, and identify polyfunctional gene states.
This is a fundamentally different way of analyzing biologica data. Genes don't have fixed semantic. Their functions emerge from context: the cellular state, tissue environment, developmental stage, and physiological condition. Only by learning genotype and phenotype together can we capture this dynamic, contextual reality.
Conclusion
The genotype-phenotype relationship is one of biology's deepest puzzles. For over a century, we've sought to map how genetic information gives rise to the stunning diversity of life. With integrated genetics and foundation models like PolyGene, we're finally building tools that match the complexity of the question.
By treating biological data like natural language and learning genes in their full phenotypic context, we unlock new dimensions of understanding: context-dependent gene functions, polyfunctional gene roles, tissue-specific networks, and systemic biomarkers that span the body.
This work lays the foundation for context-aware genomics models that understand how they emerge in the context; from basic biology to personalized medicine and designing targeted therapies.
Read the full paper: https://doi.org/10.1038/s43588-024-00765-7